
Nowe narzędzie informatyczne do analizy danych i wizualizacji wyników z metody RT-qPCR opracowane przez zespół naukowy z Wydziału Biologii z Uniwersytetu Jagiellońskiego w Krakowie.
Ilościowa reakcja łańcuchowa polimerazy w czasie rzeczywistym (ang. real time quantitative polymerase chain reaction, qPCR) to metoda analityczna szeroko stosowana zarówno w badaniach naukowych jak i diagnostyce. W metodzie tej poziom ekspresji badanych genów analizowany jest względem genu/genów referencyjnych, czyli takich, których poziom ekspresji powinien być stały niezależnie od czynników środowiskowych jakim podlega komórka lub jej stanu fizjologicznego. Istotnym wyzwaniem, a równocześnie pewnym ograniczeniem jest właściwy wybór genów referencyjnych. Arbitralny ich dobór np. jedynie na podstawie literatury, skutkuje błędnym wyznaczeniem poziomu ekspresji genów badanych, a co za tym idzie błędną interpretacją wyników. Z kolei stosowanie ogólnie dostępnych programów komputerowych do wyboru referencji, np. geNorm, NormFinder czy BestKeeper prowadzi do uzyskania różnych wyników, co związane jest z ograniczeniami jakie te algorytmy ze sobą niosą.
Próbując rozwiązać ten problem zespół naukowy z Wydziału Biologii Uniwersytetu Jagiellońskiego w składzie dr hab. Dorota Hoja-Łukowicz, prof. UJ oraz dr Marcelina Janik, opracował narzędzie informatyczne do analizy danych i wizualizacji wyników z metody RT-qPCR działające w oparciu o nowatorską metodę uzyskiwania ulepszonej referencji w połączeniu z metodą określenia spójności poziomu ekspresji danego genu badanego w modelu eksperymentalnym oraz w szeregu podmodeli, pełniących funkcję modeli pomocniczych (Rys. 1).
Omawiane narzędzie informatyczne generuje podmodele z próbek wchodzących w skład modelu eksperymentalnego, przy czy najmniejsza liczba próbek w podmodelu to dwie. Następnie, przy użyciu zaimplementowanego algorytmu NormFinder, w każdym z tych modeli (model eksperymentalny oraz modele pomocnicze) typowany jest najlepszy gen lub para genów referencyjnych z grupy analizowanych w danym eksperymencie potencjalnych genów referencyjnych. Wytypowany w ten sposób gen lub para genów będących najlepszą referencją w badanym modelu stanowi następnie podstawę do przeprowadzenia dalszych etapów analizy, tj. walidacji spójności wyników otrzymanych w oparciu o wytypowane geny referencyjne. Obliczane jest zatem RQ dla genów badanych i w ramach każdego z modeli prowadzone są analizy statystyczne oparte na odpowiednio dobranych testach statystycznych, pokazujące istotne statystyczne różnice w ekspresji badanego genu w obrębie danego modelu. Kolejnym etapem prowadzonej analizy jest porównanie spójności otrzymanych wyników/wartości i ich statystycznej istotności dla danego genu badanego w obrębie wszystkich badanych modeli. Przygotowany program wyznacza tzw. coherence score (współczynnik spójności; CS) opisujący poziom spójności otrzymanych wyników. Osiągnięcie niezadowalającej wartości współczynnika spójności, poniżej akceptowalnej przez badacza (domyślnie poniżej wartości 1), prowadzi do kolejnego etapu analizy, tj. ponownego wyboru genów referencyjnych we wszystkich badanych modelach, ale odbywa się to po uprzednim usunięciu z listy potencjalnych genów referencyjnych tego, który na poprzednim etapie uzyskał najsłabszy poziom stabilności w danym modelu (czyli np. z grupy wyjściowych n genów referencyjnych usuwany jest ten o najsłabszym wyniku stabilności i właściwy gen referencyjny jest wybierany spośród pozostałych n-1 potencjalnych genów referencyjnych). Dalej analiza prowadzona jest analogicznie jak poprzednio i zostaje zakończona przypisaniem współczynnika spójności dla genu badanego. Tym razem wartość współczynnika spójności jest obliczana na podstawie porównania wyników dla danego genu badanego w obu przeprowadzonych do tej pory etapach analizy (porównanie wyników obliczonych we wszystkich modelach na podstawie genu referencyjnego wybranego spośród n i spośród n-1 potencjalnych genów referencyjnych). Przy powtarzającym się braku wymaganego poziomu spójności analiza powtarzana jest kolejny raz po usunięciu we wszystkich modelach kolejnego potencjalnego genu referencyjnego o najsłabszym wyniku stabilności, tym razem spośród n-1 potencjalnych genów referencyjnych. Na każdym z etapów usuwania genu o najsłabszej stabilności, narzędzie generuje pliki informujące o otrzymanych wynikach analiz, przedstawionych w formie tabel oraz ostatecznie w formie wykresów pudełkowych (ang. box-plot), z naniesionymi na wykres informacjami o istotności statystycznej wyników (Rys. 2). Rozwiązanie łączy w sobie funkcjonalności programów wyznaczających najlepsze geny referencyjne, oraz programów typu arkuszy kalkulacyjnych, pozwalających na analizę poziomu ekspresji genu badanego, analizę statystyczną otrzymanych wyników i ich interpretację graficzną. Istotnym elementem rozwiązania jest zaimplementowanie określonego schematu analizy danych RT-qPCR (usuwanie genów o najsłabszej stabilności z następującą po tym ponowną analizą danych surowych) oraz wprowadzenie wartości współczynnika spójności określającego rzetelność/wiarygodność przeprowadzonej analizy. Usprawnieniem całości procesu jest także automatyczne generowanie wykresów pudełkowych prezentujących otrzymane wyniki analizy, włącznie z zaznaczonymi na wykresie istotnościami statystycznymi pomiędzy badanymi elementami danego modelu. Program umożliwia jednoczesną, niezależną analizę wielu genów badanych.
Rys. 1
Schemat przedstawiający etapy analizy danych RT-qPCR: wyznaczenie najlepszych genów referencyjnych dla każdego modelu, analiza względnej ekspresji genu badanego, analiza statystyczna i określenie poziomu spójności otrzymanych wyników w analizowanych modelach.

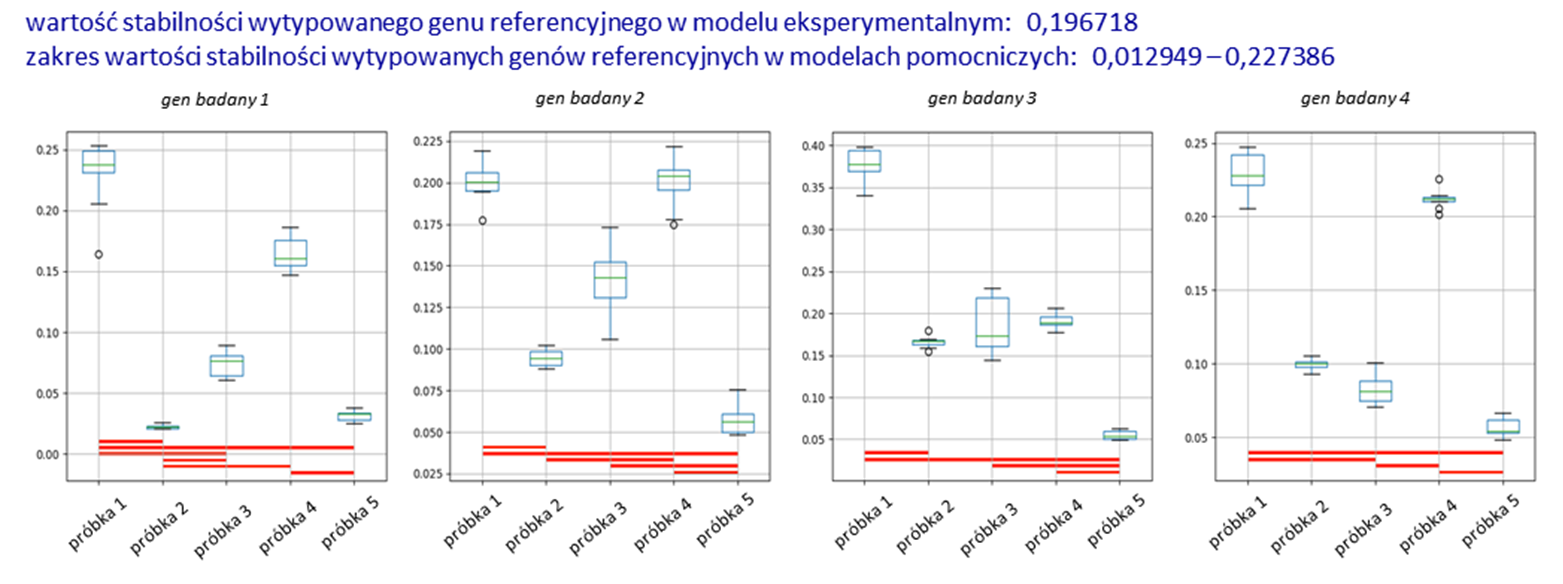
Rys. 2
Przykładowy wynik analizy.
Opracowane narzędzie informatyczne umożliwia:
1) selekcję referencji, o ulepszonej wartości stabilności, z wyjściowej puli potencjalnych genów referencyjnych;
2) określenie rzeczywistego poziomu ekspresji genów badanych i
3) prawidłową interpretację biologiczną wyników na podstawie kompleksowej analizy danych surowych otrzymanych w reakcji RT-qPCR dla wieloelementowych modeli eksperymentalnych (duże grupy badawcze złożone z linii komórkowych, próbek pobranych od pacjentów lub zwierząt).
Obecnie Centrum Transferu Technologii CITTRU UJ poszukuje podmiotów zainteresowanych współpracą przy wdrażaniu wynalazku.
Program do pobrania TUTAJ.
Narzędzie informatyczne zostało opracowane z funduszy grantowych Narodowego Centrum Nauki, Polska (projekt badawczy nr 2016/21/B/NZ3/00348).
